Genetics of Dupuytren’s Disease
Paul M.N. Werker 2
1 Department of Plastic and Reconstructive Surgery, Oxford University Hospitals NHS Trust, Nuffield Orthopaedic Centre, Oxford, United Kingdom
2 Department of Plastic Surgery, University of Groningen and University Medical Centre Groningen, Groningen, Netherlands
Abstract
It was first recognised in 1833 that there was an inherited predisposition to Dupuytren’s Disease, before DNA and genes were discovered [1]. Since then, hand surgeons have continued to be fascinated by the nature and extent of this predisposition. As epidemiological methodology and molecular genetic techniques have developed, our understanding has advanced, such that the design of novel therapeutic interventions based on this knowledge is becoming a realistic possibility.
In this section, we begin by briefly reviewing important principles and concepts of genetic inheritance. We then assess the evidence for a genetic predisposition to Dupuytren’s Disease. Next we consider the molecular genetic studies that have helped define the magnitude and nature of that genetic predisposition. Finally, we consider potential future developments in this fast developing field.
The basics of genetic inheritance
Introduction
Genetic information is passed on from generation to generation via the DNA. DNA is a complex molecule organised into the famous double helix configuration, made up of two twisting paired strands. The information passed on from generation to generation is encoded in the bases of this complex molecule – named Guanine, Cytosine, Adenine, and Thymine (G, C, A, T) – and it is the order of these bases along the DNA molecule that makes up the human genome sequence. Each base is paired with a consistent partner on the other strand of DNA - A pairs with T, and G pairs with C. The DNA is organised into chromosomes, and each of us has 22 pairs of “autosomes”, and either two X chromosomes or an X chromosome and a Y chromosome, which determine if we are female or male respectively. We inherit one copy of each chromosome from our mother and one from our father at fertilisation, and our complete genome is present in all of the cells of our body, except spermatozoa and oocyts. This means that we each have two copies (called alleles) of every gene, one from each parent.
In total, the human genome consists of around three billion base pairs. Some of this DNA sequence is transcribed and translated within our cells to make proteins. These regions of DNA are called protein-coding genes. In total, humans have around 24,000 protein coding genes. In fact, these genes only make up around 1.5% of the entire DNA contained in our cells. The rest of the genome consists of “non-coding” DNA. This non-coding DNA has important functions, including the highly complex spatial and temporal regulation of expression of protein coding genes.
Before DNA was discovered, an Austrian monk named Gregor Mendel established the principles of inheritance of genes [2]. He experimented on pea plants, cross-fertilising those with opposing characteristics – tall with short, smooth with wrinkled – and also their offspring. His results established the law of segregation, that there are dominant and recessive traits passed on from parent to offspring (see Box 1). As such, he is considered to be the founding father of modern genetics.
What is normal variation, and what is a mutation?
Within the three billion base pairs of our genome, around 30 million base pairs are what is known as polymorphic. This means they exist in two or more forms within the normal population. This is the normal variation that makes each of us different from the other. A minority of these variants are within protein coding genes, and will have a direct effect of the function of that gene. However, the vast majority of these variants are within non-coding DNA, and have subtle effects within the cell, such as changing the expression of a protein-coding gene. Some of these effects will be beneficial, and some deleterious, to the survival and reproductive fitness of the individual. Such variants will be retained or lost, respectively, within the population over time. This is the basis of Darwin’s theory of natural selection.
In genetic terms, a mutation is a change in the DNA sequence. This may take the form of a change in a single base pair, or the deletion, insertion, or rearrangement of larger parts of DNA, or even a whole chromosome. A mutation generally has the property of being disadvantageous to the organism. Most mutations are found within the protein coding genes, and lead to disruption of either the whole of the protein that the gene encodes, or a vital part of the protein required for its function. Diseases caused by such mutations tend to be inherited in regular predictable patterns – Autosomal Dominant, Autosomal Recessive, and X-linked recessive, see Box 1 – and are termed Mendelian diseases in honour of Gregor Mendel. Because the effects of these mutations are deleterious, their frequency within the population is usually very low. Furthermore, the syndromes caused by such mutations are usually (though not exclusively) present and recognised at birth or in childhood.
Mendelian diseases within hand surgery
Hand surgery occupies a special place in the history of human genetics, as the very first human disease shown to follow an autosomal dominant Mendelian pattern of inheritance was brachydactyly type A1. Subsequently, this has been shown to be caused by mutations in the gene Indian Hedgehog (IHH) [3]. Most congenital hand malformation syndromes display autosomal dominant inheritance. A rare example of an autosomal recessive hand surgery condition is one of the variants of split-hand/split- foot malformation, characterised by central clefts of the hands and feet. This has been shown to be caused by mutations in the gene WNT10B [4], [5], [6], [7]. X-linked recessive congenital hand malformations are extremely rare, an example being a family with fusion of the 4th and 5th metacarpals [8].
However, Mendelian inheritance is not always clear-cut. For example, not all people who carry a particular dominant mutation will show signs of the disease on clinical examination. This phenomenon is known as incomplete penetrance. For example, a mutation within a regulatory region of the gene Sonic Hedgehog has been shown to cause triphalangeal thumb [9]. In this study, only around half of people who carried the mutation were found to actually have triphalangeal thumb on clinical examination. Also, patients with precisely the same mutation in the same gene may have a slightly different phenotype. This is known as variable expressivity. These phenomena are due to the modifying effects of other variants within the genome, the effects of the environment, or to random effects.
What is a complex disease?
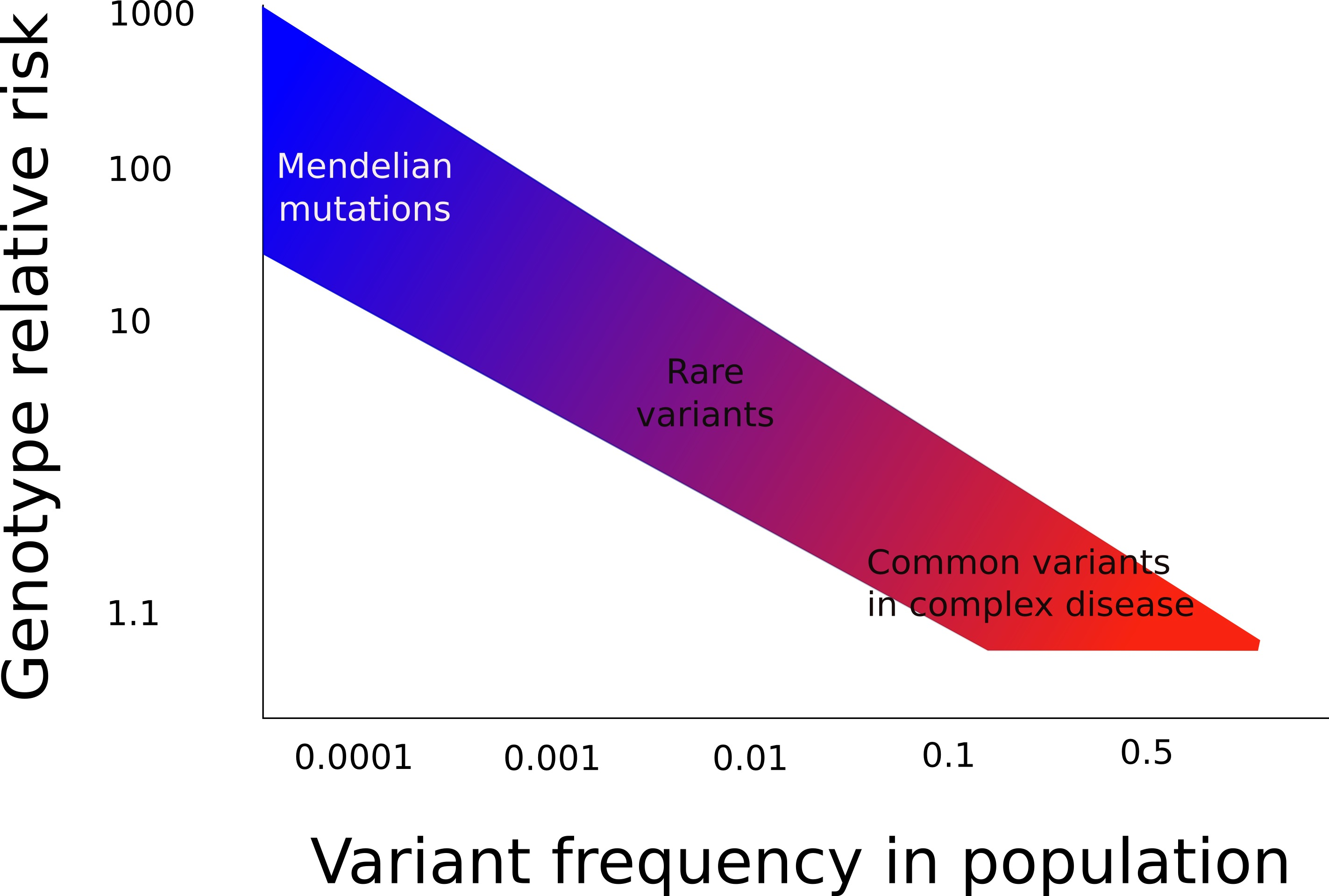
As a consequence of the above, it is helpful to consider variants and mutations as lying on a spectrum (Figure 1). At the one end are mutations that are very rare, but have a very large increased risk of causing a particular disease within an organism. At the other end are variants that are very common, but have a very small increased risk of causing a particular disease within an organism. For Mendelian diseases, only rare mutations in a single gene with a high effect size cause the disease.
However, there are many diseases, for example heart disease and rheumatoid arthritis, whereby a familial predisposition is well recognised, but mutations in single genes are not responsible for this predisposition, and the inheritance does not follow simple Mendelian patterns. Here, multiple common variants each slightly increase the risk of a person getting the disease. These variants are often in non-coding areas of the DNA, and exert their effects by subtle changes in gene function. Furthermore, they interact with environmental influences, and this interaction determines the final expression of the disease. These diseases are known as complex diseases, and discovering the basis of the genetic predisposition is more difficult than for Mendelian diseases.
When considering complex diseases, it is helpful to think of a threshold effect (Figure 2). Here, many genetic variants each increase a person’s chance of getting a particular disease by a small amount. The more of these predisposing variants that an individual carries, the closer they are to tipping over the threshold, and displaying the disease. Environmental factors interact with these genetic factors to provide a further push, leading to the presence of the disease in an individual. If there is a greater genetic predisposition (a stronger family history), more predisposing genetic factors are present, and less environmental factors are required to cause the disease to become manifest. Similarly, a person with little genetic predisposition can still be pushed over the threshold and express the disease by having multiple excessive environmental exposures.
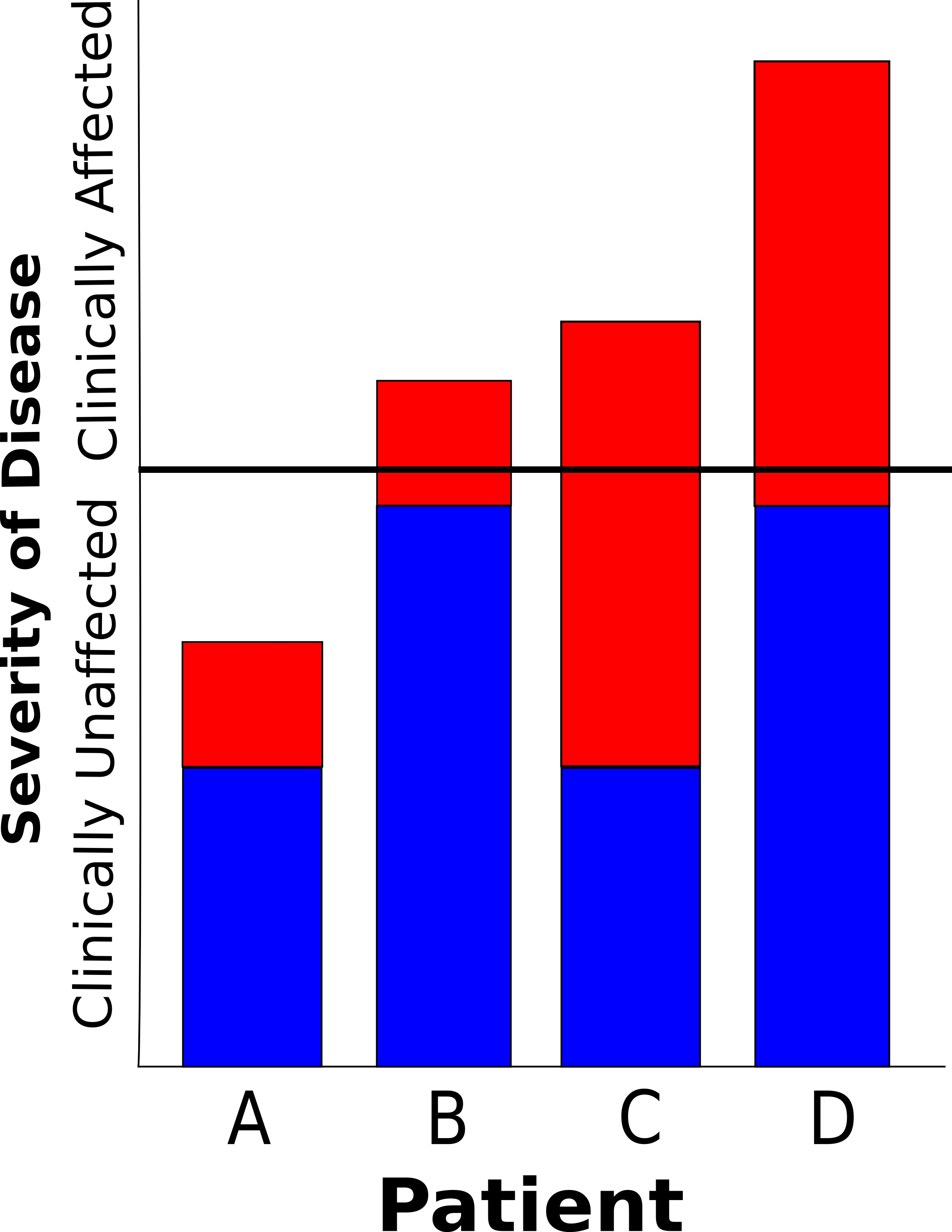
A further consequence of this model, is that people with a larger genetic predisposition tend to have earlier onset disease – they need less environmental insult to push them over the threshold – and the disease tends to be more severe – they get further over the threshold with a similar amount of environmental exposure to a person who has a smaller genetic predisposition.
When considering this threshold model, it becomes clear that Dupuytren’s Disease is very typical of a complex disease, just like heart disease, diabetes, and rheumatoid arthritis. In Dupuytren’s Disease we observe a condition that has a familial tendency, but doesn’t follow simple Mendelian inheritance rules. The onset of the disease is in later life, and earlier onset disease tends to have a stronger family history than later onset disease. Patients with a strong family history also tend to be more severely affected. Furthermore, there are clear and well documented environmental influences on the expression of the disease. As shown towards the end of the chapter, this concept of Dupuytren’s Disease being a complex disease has proved to be correct, and we are now beginning to understand some of the genetic variants that cause the predisposition. We hope that this will form the basis for the rational design of new therapies aimed at preventing both the disease in the first place, and also recurrence after treatment in high-risk individuals.
The evidence for a genetic predisposition to Dupuytren’s Disease
Population studies
Modern genetic studies have defined the frequency of variants throughout the genome in different populations. This has confirmed the hypothesis that populations from different parts of the world have distinguishable genetic backgrounds. If a disease is more common in particular racial groups, then a genetic predisposition to that particular disease is possible. Many studies have evaluated the prevalence of Dupuytren’s Disease within a particular country (reviewed in [10]). Estimates of prevalence vary widely depending on the assessor, diagnostic criteria, and co-morbid conditions of the population under study.
When considering studies not designed to evaluate a cohort with a specific risk factor for Dupuytren’s Disease, the prevalence in Scandinavian countries varies from 6% to 11% [11], [12] , [13]; in Iceland it is reported to be between 13% and 19% [14], [15]; in the UK 3.5% to 30% [16], [17], [18], [19], [20], [21]; in France and Spain 8% to 9% [22], [23]; in North America, the prevalence is 3.4% to 33% [24], [25].
In the Netherlands, the overall community prevalence was 22.1%, but those with contracture were only 4.2% of the study population [26]. Similarly, in Belgium, the population prevalence was 32%, but only 8% of males and 4% of females had contracture [27]. These results suggest a generally benign natural history of the disease, though population-based longitudinal studies would be required to confirm this. Interestingly, the documented prevalence in India is also 4% [28]. Dupuytren’s Disease among black Africans is limited to case reports [29].
Of course, populations also share environmental influences, so stronger evidence that genetics is important comes from studies that demonstrate a different incidence in different racial groups within the same environment, or demonstrate that the incidence of a disease persists when the population moves to a different environment. Finsen and co-authors found that the prevalence of Dupuytren’s Disease amongst the native Sami of northern Scandinavia was 5.8%, and in Norwegians living in the same area was 10.1% [30]. Though this difference was not statistically significant, it does suggest that genetically distinct populations have different prevalence of disease. Furthermore, Heuston studied the prevalence of Dupuytren’s Disease in Australia, a country populated by people from northern Europe who have migrated to a different environment [31]. He found a prevalence rate of 23%, equivalent or higher than that found in Europe.
Overall, Dupuytren’s is widely considered to be a disease of Europeans, and more specifically Northern Europeans, but large variability in the data make this conclusion less secure than is generally assumed. Detailed genetic studies should provide a more definitive answer to this question in the coming years.
Twin and family studies
Studying twins is a powerful way to document and quantify the magnitude of genetic risk in a disease. In twin studies, concordance for a particular disease is defined as twins having the same disease status – i.e. both twins have the disease or both twins do not have the disease – whereas discordance is defined as twins not having the same disease status – i.e. one twin has the disease and the other does not. Monozygotic (identical) twins share their entire genome, whereas dizygotic (non-identical) twins, like normal siblings, share on average 50% of their genome. Thus, if monozygotic twins are more commonly concordant for a particular disease than dizygotic twins, this is good evidence that genetics play a part in the predisposition to disease.
In Dupuytren’s Disease, no systematic review of twin registries has been published to date. The data on twin concordance is limited to case reports, and shows variable effects, with some authors reporting concordant twin pairs [32], and others discordant twin pairs [33], [34].
Several authors have attempted to document and quantify the familial incidence of Dupuytren’s Disease. In the UK, 7–15% of those with Dupuytren’s Disease reported a family history of the disease [35], [36], and in France the corresponding figure was 20% [23]. However, many people with early Dupuytren’s Disease may not recognise that they are affected, and certainly their relatives may be unaware of them having the condition. It is therefore likely that these studies underestimate the true incidence of a positive family history. Indeed, Ling obtained a positive family history of Dupuytren’s Disease in 8/50 patients but discovered that 34/50 patients actually had a relative with Dupuytren’s Disease when he personally examined 830 of their relatives [37].
A standard method of quantifying familial risk to disease is the sibling recurrence risk. This compares the rate of the disease under study in the siblings of index cases, compared to the rate in the general population. Because siblings share on average 50% of their DNA, a higher prevalence in siblings is strong evidence for a genetic predisposition. For example, in rheumatoid arthritis, a sibling recurrence risk of between 3 and 7 has been reported, with higher values for more severe disease [38], and in anteromedial osteoarthritis of the knee, the sibling recurrence risk has been calculated to be 3.21 [39].
In Dupuytren’s Disease, the sibling recurrence risk has been calculated in the UK. The authors examined a randomly selected single sibling of 100 Dupuytren’s Disease index cases, and compared the rate of Dupuytren’s Disease to that of 124 controls ascertained from an ophthalmology clinic for non-diabetic eye conditions. They found that 47% of siblings were affected compared to 10.5% of controls, giving a sibling recurrence risk of 4.5 [40]. Furthermore, using a history of surgery for Dupuytren’s Disease as a proxy marker of disease severity, they found the sibling recurrence risk for surgery to be 7.1, suggesting more severe disease is more highly heritable, consistent with the threshold model of complex disease presented above (Figure 2).
Molecular studies
Linkage studies
In single gene disorders, large families where a disease is passing from generation to generation can be used to determine where on the chromosomes a genetic mutation using molecular techniques of genetic linkage. Here, genetic markers are typed across the entire genome in affected and unaffected family members, and segments of chromosomes where the variants are similar in affected individuals, but different in unaffected individuals are said to be “linked” to the disease. This technique works well for Mendelian disease, where rare mutations have a large effect (Figure 2) and for decades was the bedrock for discovering disease-causing mutationssease genes for several decades. However, for complex diseases linkage cannot detect common variants that predispose to disease [41], [42].
It is possible, in a small minority of cases, that a rare variant in a gene is predisposing to disease with a high effect size in a single family, or several closely related families, effectively acting like a mutation. Such predisposition could be studied by linkage analysis, and there is a single report of this in the literature for Dupuytren’s Disease [43]. Here, researchers performed a linkage analysis on a large five-generation Swedish family, and found evidence of linkage to a region on chromosome 16 that just reached statistical significance. This result has not been confirmed since the original publication.
Association studies
Genetic association studies test for a correlation between disease status and genetic variants in cases and controls. A higher frequency of an allele (see introduction) in cases compared to controls suggests that the tested variant, or another untested variant that is close to the tested variant, predisposes to disease.
There are two important principles in association studies. Firstly, any variant found to be associated with a complex disease should be replicated in an independent cohort of cases and controls. Secondly, when multiple different variants are tested for association, the p-value accepted as statistically significant should be appropriately lowered by a Bonferroni correction [44]. These measures help to guard against statistical false positives.
Association studies can be divided into candidate gene studies and genome-wide association studies (GWAS).
Candidate gene studies
Before the advent of molecular genetic techniques that enabled researchers to study variation across the entire genome, researchers hypothesized which of the approximately 24,000 protein-coding genes might be involved in a particular complex disease. They found a small minority of the hundreds of variants within the gene, and tested them for association with the disease under study, typically using tens or a few hundred cases and controls. As will be evident from the above discussion of human variation, the chance of correctly identifying a predisposing variant in this way is small.
Furthermore, experience from GWAS (see below), has demonstrated that the majority of potential candidate gene associations reported across the literature in a wide variety of complex diseases, do not stand up to rigorous independent replication [45], [46], [47].
A similar picture has emerged with Dupuytren’s Disease. Associations between variants in many different genes have been investigated in small sample groups. These studies often also have methodological flaws, such as reporting statistical significance without performing a Bonferroni correction for multiple hypothesis testing [48]. Some positive associations have been reported [48], [49], [50], [51], [52], [53]. Some negative associations have also been reported [54], [55], [56], [57]. Of note, none of the positive associations have been replicated in independent cohorts, or in the GWAS described below.
Genome-wide association studies (GWAS)
The current gold-standard technique for studying the genetics of complex disease is the GWAS. Here, hundreds of thousands or millions of variants across the entire genome are typed in cases and controls, and tested for association with the disease. The biggest advantage of this technique is that it does not require any pre-existing knowledge of the pathogenesis of the disease in question: all areas of the genome are interrogated for association with the disease and assessed equally. This has led to the discovery of associations of genes with diseases that were previously unsuspected, leading to new avenues of research into disease pathogenesis. Indeed, the majority of robust associations that have been discovered are not within protein-coding genes at all, but non-coding DNA, and are likely to regulate gene expression [58].
However, the vast number of statistical tests performed in a GWAS leads to an unprecedented potential for false-positive results. Thus an adequate Bonferroni correction must be performed, typically leading to P<0.00000005 being accepted as statistical evidence for association. This means that thousands of cases and controls must be tested for association in order to achieve significance [59], [60].
The GWAS approach has proved extremely successful in the search for variants predisposing to complex disease, with over 15,000 associations reported in the National Human Genome Research Institute catalogue of published GWAS [61]. Whilst the direct application of these results to clinical medicine has proved to be frustratingly slow [62], progress towards clinical application is being made in diseases such as rheumatoid arthritis [63].
In Dupuytren’s Disease, a European GWAS has been reported [64]. In phase 1 of the study, the authors genotyped 960 Dutch patients with Dupuytren’s Disease, and 3,117 controls patients in a discovery cohort. They then attempted replication of the significant results from phase 1 in an independent replication cohort consisting of 1,365 cases and 8,445 controls from the UK, Germany and Holland. The results of this analysis revealed nine genetic regions that pre-dispose to Dupuytren’s Disease. None of these associated variants were in protein-coding genes, and none of them were close to any of the reported associations from previous candidate gene association studies.
The authors went on to perform a bioinformatic analysis of the protein coding genes that were close to the variants associated with Dupuytren’s Disease. This revealed a significant excess of genes involved in the WNT signaling pathway. This suggests that WNT signaling may be important in the pathogenesis of Dupuytren’s Disease, though this observation requires further investigation.
The results of this GWAS have been used to further explore the genetics of Dupuytren’s Disease. Calculations based on the sibling recurrence risk reveal that the genetic regions discovered in this study account for around 12% of the genetic predisposition to Dupuytren’s Disease, implying that larger studies with more cases and controls will discover many more associated variants [40]. Furthermore, a positive correlation has been described between increasing numbers of genetic risk variants and early onset disease, positive family history, and features of a Dupuytren’s diathesis [65]. Again, this data is consistent with the threshold model presented in figure 2. A further independent study has confirmed that the most statistically significantly associated variant is associated with Dupuytren’s Disease in a Polish population [66].
Future perspectives
Further genetic studies
Molecular genetic technology continues to evolve at a rapid pace. It is evident from studies of other complex diseases that studying larger case-control cohorts leads to the discovery of further common variants that pre-dispose to disease with smaller effect size [67], and these variants can give valuable insights into the biology of the disease. This principle is currently being applied to Dupuytren’s Disease, with a much larger GWAS currently being undertaken in European populations.
When the first draft of the human genome was published in 2001, it was estimated that the cost of this sequence was around $100 million. It is now possible to sequence an entire human genome for under $7,000, and the cost is still falling [68]. We expect that genetic sequence data will translate into routine clinical practice in many areas over the coming decades. We therefore also expect that Dupuytren’s Disease patients will undergo whole genome sequencing, and their sequence data will be compared to controls without Dupuytren’s Disease, in order to discover the whole range of genetic variation, both common and rare variants, that predispose to disease. As an intermediate step, it is possible to sequence just the protein-coding genes within the genome – so-called exome sequencing. This technique has proved excellent for discovering mutations causing Mendelian disease, and may also find some application in complex diseases [69], [70].
However, there are currently massive barriers to the storage, analysis, and interpretation of both exome sequencing and whole-genome sequencing data. The development of bioinformatic techniques has lagged behind molecular developments, and training researchers in this area will be the key to further advances in the field.
Implications for treatment
The goal of genetic research into Dupuytren’s Disease is to understand the pathophysiology of the disease, leading to the rational design of new treatments aimed at preventing development of the disease, and preventing recurrence after surgical or other treatment. Furthermore, in the future it is possible that genetic risk data will be taken into account when planning surgery. For example, a patient with a high risk of recurrence based on genetic and clinical data might require a more extensive primary procedure.
These goals are realistic in the medium to long term, but many challenges remain. As surgeons, we have unique access to patients with this fascinating disease. We should strive to improve the treatment of current patients by recruiting to high quality randomized controlled trials of current treatments, and strive to improve the treatment of future patients by recruiting to high quality basic and applied science research into the disease.
References
[1] Goyrand G. Nouvelles recherches sur la rétraction permanente des doigts. Mem R Méd Belg. 1833;3:489.[2] Mendel G. Versuche über Pflanzen-Hybriden. Verhandlungen des Naturforschenden Vereines in Brünn. 1866;4:3-47.
[3] Gao B, Guo J, She C, Shu A, Yang M, Tan Z, Yang X, Guo S, Feng G, He L. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat Genet. 2001 Aug;28(4):386-8. DOI: 10.1038/ng577
[4] Aziz A, Irfanullah, Khan S, Zimri FK, Muhammad N, Rashid S, Ahmad W. Novel homozygous mutations in the WNT10B gene underlying autosomal recessive split hand/foot malformation in three consanguineous families. Gene. 2014 Jan;534(2):265-71. DOI: 10.1016/j.gene.2013.10.047
[5] Blattner A, Huber AR, Röthlisberger B. Homozygous nonsense mutation in WNT10B and sporadic split-hand/foot malformation (SHFM) with autosomal recessive inheritance. Am J Med Genet A. 2010 Aug;152A(8):2053-6. DOI: 10.1002/ajmg.a.33504
[6] Khan S, Basit S, Zimri FK, Ali N, Ali G, Ansar M, Ahmad W. A novel homozygous missense mutation in WNT10B in familial split-hand/foot malformation. Clin Genet. 2012 Jul;82(1):48-55. DOI: 10.1111/j.1399-0004.2011.01698.x
[7] Ugur SA, Tolun A. Homozygous WNT10b mutation and complex inheritance in Split-Hand/Foot Malformation. Hum Mol Genet. 2008 Sep;17(17):2644-53. DOI: 10.1093/hmg/ddn164
[8] Lonardo F, Della Monica M, Riccardi G, Riccio I, Riccio V, Scarano G. A family with X-linked recessive fusion of metacarpals IV and V. Am J Med Genet A. 2004 Feb 1;124A(4):407-10.
[9] Furniss D, Lettice LA, Taylor IB, Critchley PS, Giele H, Hill RE, Wilkie AO. A variant in the sonic hedgehog regulatory sequence (ZRS) is associated with triphalangeal thumb and deregulates expression in the developing limb. Hum Mol Genet. 2008 Aug;17(16):2417-23. DOI: 10.1093/hmg/ddn141
[10] Hindocha S, McGrouther DA, Bayat A. Epidemiological evaluation of Dupuytren's disease incidence and prevalence rates in relation to etiology. Hand (N Y). 2009 Sep;4(3):256-69. DOI: 10.1007/s11552-008-9160-9
[11] Mikkelsen OA. The prevalence of Dupuytren's disease in Norway. A study in a representative population sample of the municipality of Haugesund. Acta Chir Scand. 1972;138(7):695-700.
[12] Bergenudd H, Lindgarde F, Nilsson BE. Prevalence of Dupuytren's contracture and its correlation with degenerative changes of the hands and feet and with criteria of general health. J Hand Surg Br. 1993;18:254-7. DOI: 10.1016/0266-7681(93)90123-W
[13] Finsen V, Dalen H, Nesheim J. The prevalence of Dupuytren's disease among 2 different ethnic groups in northern Norway. J Hand Surg Am. 2002 Jan;27(1):115-7. DOI: 10.1053/jhsu.2002.29486
[14] Gudmundsson KG, Arngrimsson R, Sigfusson N, Jonsson. Prevalence of joint complaints amongst individuals with Dupuytren's disease – from the Reykjavik study. Scand J Rheumatol. 1999;28:300-4. DOI: 10.1080/03009749950155481
[15] Gudmundsson KG, Arngrimsson R, Sigfusson N, Bjornsson A, Jonsson T. Epidemiology of Dupuytren's disease: clinical, serological, and social assessment. The Reykjavik Study. J Clin Epidemiol. 2000;53:291-6. DOI: 10.1016/S0895-4356(99)00145-6
[16] Herzog EG. The aetiology of Dupuytren's contracture. Lancet. 1951;1:1305-6. DOI: 10.1016/S0140-6736(51)91775-8
[17] Early PF. Population studies in Dupuytren's contracture. J Bone Joint Surg Br. 1962;44:602-13. DOI: 10.1302/0301-620X.44B3.602
[18] Mackenney RP. A population study of Dupuytren's contracture. Hand. 1983;15:155-61. DOI: 10.1016/S0072-968X(83)80007-2
[19] Lennox IA, Murali SR, Porter R. A study of the repeatability of the diagnosis of Dupuytren's contracture and its prevalence in the grampian region. J Hand Surg Br. 1993;18:258-61. DOI: 10.1016/0266-7681(93)90124-X
[20] White HA, Alcolado R, Alcolado JC. Examination of the hands: an insight into the health of a Welsh population. Postgrad Med J. 2003 Oct;79(936):588-9. DOI: 10.1136/pmj.79.936.588
[21] Burke FD, Proud G, Lawson IJ, McGeoch KL, Miles JN. An assessment of the effects of exposure to vibration, smoking, alcohol and diabetes on the prevalence of Dupuytren's disease in 97,537 miners. J Hand Surg Eur Vol. 2007 Aug;32(4):400-6. DOI: 10.1016/J.JHSE.2005.02.002
[22] Quintana Guitian A. [Various epidemiologic aspects of Dupuytren's disease]. Ann Chir Main. 1988;7(3):256-62. DOI: 10.1016/S0753-9053(88)80013-9
[23] Lucas G, Brichet A, Roquelaure Y, Leclerc A, Descatha A. Dupuytren's disease: personal factors and occupational exposure. Am J Ind Med. 2008 Jan;51(1):9-15. DOI: 10.1002/ajim.20542
[24] Gordon S. Dupuytren's contracture: the significance of various factors in its etiology. Ann Surg. 1954 Nov;140(5):683-6. DOI: 10.1097/00000658-195411000-00007
[25] Yost J, Winters T, Fett HC Sr. Dupuytren's contracture; a statistical study. Am J Surg. 1955 Oct;90(4):568-71.
[26] Lanting R, van den Heuvel ER, Westerink B, Werker PM. Prevalence of Dupuytren disease in The Netherlands. Plast Reconstr Surg. 2013 Aug;132(2):394-403. DOI: 10.1097/PRS.0b013e3182958a33
[27] Degreef I, De Smet L. A high prevalence of Dupuytren's disease in Flanders. Acta Orthop Belg. 2010 Jun;76(3):316-20.
[28] Dasgupta AK, Harrison J. Effects of vibration on the hand-arm system of miners in India. Occup Med (Lond). 1996 Feb;46(1):71-8. DOI: 10.1093/occmed/46.1.71
[29] Mennen U. Dupuytren's contracture in the Negro. J Hand Surg Br. 1986 Feb;11(1):61-4.
[30] Finsen V, Dalen H, Nesheim J. The prevalence of Dupuytren's disease among 2 different ethnic groups in northern Norway. J Hand Surg Am. 2002 Jan;27(1):115-7. DOI: 10.1053/jhsu.2002.29486
[31] Hueston JT. Further studies on the incidence of Dupuytren's contracture. Med J Aust. 1962 Apr;49(1):586-8.
[32] Couch H. Identical dupuytren's contracture in identical twins. Can Med Assoc J. 1938 Sep;39(3):225-6.
[33] Lyall HA. Dupuytren's disease in identical twins. J Hand Surg Br. 1993 Jun;18(3):368-70. DOI: 10.1016/0266-7681(93)90066-O
[34] Beleta H, Forés J. Dupuytren's disease in a rock climber with an unaffected identical twin. J Hand Surg Eur Vol. 2014 Mar;39(3):313-4. DOI: 10.1177/1753193412457149
[35] Kelly SA, Burke FD, Elliot D. Injury to the distal radius as a trigger to the onset of Dupuytren's disease. J Hand Surg Br. 1992 Apr;17(2):225-9. DOI: 10.1016/0266-7681(92)90096-K
[36] Smith SP, Devaraj VS, Bunker TD. The association between frozen shoulder and Dupuytren's disease. J Shoulder Elbow Surg. 2001 Mar-Apr;10(2):149-51.
[37] Ling RS. The genetic factor in dupuytren's disease. J Bone Joint Surg Br. 1963 Nov;45(4):709-18.
[38] Wordsworth P, Bell J. Polygenic susceptibility in rheumatoid arthritis. Ann Rheum Dis. 1991 Jun;50(6):343-6.
[39] McDonnell SM, Sinsheimer J, Price AJ, Carr AJ. Genetic influences in the aetiology of anteromedial osteoarthritis of the knee. J Bone Joint Surg Br. 2007 Jul;89(7):901-3. DOI: 10.1302/0301-620X.89B7.18915
[40] Capstick R, Bragg T, Giele H, Furniss D. Sibling recurrence risk in Dupuytren's disease. J Hand Surg Eur Vol. 2013 May;38(4):424-9. DOI: 10.1177/1753193412453359
[41] Anderson CA, Soranzo N, Zeggini E, Barrett JC. Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biol. 2011 Jan 18;9(1):e1000580. DOI: 10.1371/journal.pbio.1000580
[42] Orozco G, Barrett JC, Zeggini E. Synthetic associations in the context of genome-wide association scan signals. Hum Mol Genet. 2010 Oct;19(R2):R137-44. DOI: 10.1093/hmg/ddq368
[43] Hu FZ, Nystrom A, Ahmed A, Palmquist M, Dopico R, Mossberg I, Gladitz J, Rayner M, Post JC, Ehrlich GD, Preston RA. Mapping of an autosomal dominant gene for Dupuytren's contracture to chromosome 16q in a Swedish family. Clin Genet. 2005 Nov;68(5):424-9. DOI: 10.1111/j.1399-0004.2005.00504.x
[44] Cooper DN, Nussbaum RL, Krawczak M. Proposed guidelines for papers describing DNA polymorphism-disease associations. Hum Genet. 2002 Mar;110(3):207-8. DOI: 10.1007/s00439-001-0672-4
[45] Ricigliano VA, Umeton R, Germinario L, Alma E, Briani M, Di Segni N, Montesanti D, Pierelli G, Cancrini F, Lomonaco C, Grassi F, Palmieri G, Salvetti M. Contribution of genome-wide association studies to scientific research: a pragmatic approach to evaluate their impact. PLoS ONE. 2013;8(8):e71198. DOI: 10.1371/journal.pone.0071198
[46] Siontis KC, Patsopoulos NA, Ioannidis JP. Replication of past candidate loci for common diseases and phenotypes in 100 genome-wide association studies. Eur J Hum Genet. 2010 Jul;18(7):832-7. DOI: 10.1038/ejhg.2010.26
[47] Bevan S, Traylor M, Adib-Samii P, Malik R, Paul NL, Jackson C, Farrall M, Rothwell PM, Sudlow C, Dichgans M, Markus HS. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke. 2012 Dec;43(12):3161-7. DOI: 10.1161/STROKEAHA.112.665760
[48] Bayat A, Stanley JK, Watson JS, Ferguson MW, Ollier WE. Genetic susceptibility to Dupuytren's disease: transforming growth factor beta receptor (TGFbetaR) gene polymorphisms and Dupuytren's disease. Br J Plast Surg. 2003 Jun;56(4):328-33.
[49] Bayat A, Watson JS, Stanley JK, Ferguson MW, Ollier WE. Genetic susceptibility to dupuytren disease: association of Zf9 transcription factor gene. Plast Reconstr Surg. 2003 Jun;111(7):2133-9. DOI: 10.1097/01.PRS.0000060531.98180.32
[50] Bayat A, Walter J, Lambe H, Watson JS, Stanley JK, Marino M, Ferguson MW, Ollier WE. Identification of a novel mitochondrial mutation in Dupuytren's disease using multiplex DHPLC. Plast Reconstr Surg. 2005 Jan;115(1):134-41.
[51] Brown JJ, Ollier W, Thomson W, Bayat A. Positive association of HLA-DRB1*15 with Dupuytren's disease in Caucasians. Tissue Antigens. 2008 Aug;72(2):166-70. DOI: 10.1111/j.1399-0039.2008.01082.x
[52] Jónsson T, Gudmundsson KG, Bjarnadóttir K, Hjálmarsdótti IB, Gudmundsson S, Arngrímsson R. Association of HLA-DRB1*01 with Dupuytren's disease. Scand J Rheumatol. 2013;42(1):45-7. DOI: 10.3109/03009742.2012.713982
[53] Shih B, Tassabehji M, Watson JS, Bayat A. DNA copy number variations at chromosome 7p14.1 and chromosome 14q11.2 are associated with dupuytren's disease: potential role for MMP and Wnt signaling pathway. Plast Reconstr Surg. 2012 Apr;129(4):921-32. DOI: 10.1097/PRS.0b013e3182442343
[54] Bayat A, Alansar A, Hajeer AH, Shah M, Watson JS, Stanley JK, Ferguson MW, Ollier WE. Genetic susceptibility in Dupuytren's disease: lack of association of a novel transforming growth factor beta(2) polymorphism in Dupuytren's disease. J Hand Surg Br. 2002 Feb;27(1):47-9. DOI: 10.1054/jhsb.2001.0689
[55] Hnanicek J, Cimburova M, Putova I, Svoboda S, Stritesky J, Kratka K, Sosna B, Horak J. Lack of association of iron metabolism and Dupuytren's disease. J Eur Acad Dermatol Venereol. 2008 Apr;22(4):476-80. DOI: 10.1111/j.1468-3083.2007.02506.x
[56] Kaur S, Forsman M, Ryhänen J, Knuutila S, Larramendy ML. No gene copy number changes in Dupuytren's contracture by array comparative genomic hybridization. Cancer Genet Cytogenet. 2008 May;183(1):6-8. DOI: 10.1016/j.cancergencyto.2008.01.018
[57] Zyluk A, Debniak T, Puchalski P. Common variants of the ALDH2 and DHDH genes and the risk of Dupuytren's disease. J Hand Surg Eur Vol. 2013 May;38(4):430-4. DOI: 10.1177/1753193412471502
[58] Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA. 2008 Mar;299(11):1335-44. DOI: 10.1001/jama.299.11.1335
[59] McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008 May;9(5):356-69. DOI: 10.1038/nrg2344
[60] McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet. 2008 Oct;17(R2):R156-65. DOI: 10.1093/hmg/ddn289
[61] Hindorff LA, Morales J, Junkins HA, Hall PN, Klemm AK, Manolio TA. A Catalog of Published Genome-Wide Association Studies. Available from: https://www.genome.gov/26525384/catalog-of-published-genomewide-association-studies/
[62] Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012 Jan;90(1):7-24. DOI: 10.1016/j.ajhg.2011.11.029
[63] Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, Graham RR, Manoharan A, Ortmann W, Bhangale T, Denny JC, Carroll RJ, Eyler AE, Greenberg JD, Kremer JM, Pappas DA, Jiang L, Yin J, Ye L, Su DF, Yang J, Xie G, Keystone E, Westra HJ, Esko T, Metspalu A, Zhou X, Gupta N, Mirel D, Stahl EA, Diogo D, Cui J, Liao K, Guo MH, Myouzen K, Kawaguchi T, Coenen MJ, van Riel PL, van de Laar MA, Guchelaar HJ, Huizinga TW, Dieudé P, Mariette X, Bridges SL Jr, Zhernakova A, Toes RE, Tak PP, Miceli-Richard C, Bang SY, Lee HS, Martin J, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Rantapää-Dahlqvist S, Arlestig L, Choi HK, Kamatani Y, Galan P, Lathrop M; RACI consortium; GARNET consortiumEyre S, Bowes J, Barton A, de Vries N, Moreland LW, Criswell LA, Karlson EW, Taniguchi A, Yamada R, Kubo M, Liu JS, Bae SC, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Raychaudhuri S, Stranger BE, De Jager PL, Franke L, Visscher PM, Brown MA, Yamanaka H, Mimori T, Takahashi A, Xu H, Behrens TW, Siminovitch KA, Momohara S, Matsuda F, Yamamoto K, Plenge RM. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014 Feb;506(7488):376-81. DOI: 10.1038/nature12873
[64] Dolmans GH, Werker PM, Hennies HC, Furniss D, Festen EA, Franke L, Becker K, van der Vlies P, Wolffenbuttel BH, Tinschert S, Toliat MR, Nothnagel M, Franke A, Klopp N, Wichmann HE, Nürnberg P, Giele H, Ophoff RA, Wijmenga C; Dutch Dupuytren Study Group; German Dupuytren Study Group; LifeLines Cohort Study; BSSH-GODD Consortium. Wnt signaling and Dupuytren's disease. N Engl J Med. 2011 Jul;365(4):307-17. DOI: 10.1056/NEJMoa1101029
[65] Dolmans GH, de Bock GH, Werker PM. Dupuytren diathesis and genetic risk. J Hand Surg Am. 2012 Oct;37(10):2106-11. DOI: 10.1016/j.jhsa.2012.07.017
[66] Dębniak T, Żyluk A, Puchalski P, Serrano-Fernandez P. Common variants of the EPDR1 gene and the risk of Dupuytren’s disease. Handchir Mikrochir Plast Chir. 2013 Oct;45(5):253-7. DOI: 10.1055/s-0033-1357119
[67] Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Müller-Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stančáková A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutškov K, Langford C, Leander K, Lindholm E, Lobbens S, Männistö S, Mirza G, Mühleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurðsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvänen AC, Eriksson JG, Peltonen L, Nöthen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L; Wellcome Trust Case Control Consortium; Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) ConsortiumShuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njølstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen-Kiukaanniemi SM, Saaristo TE, Korpi-Hyövälti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jöckel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012 Sep;44(9):981-90. DOI: 10.1038/ng.2383
[68] Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). [Accessed 2014 Feb 03]. Available from: https://www.genome.gov/27541954/dna-sequencing-costs-data/
[69] Fahiminiya S, Almuriekhi M, Nawaz Z, Staffa A, Lepage P, Ali R, Hashim L, Schwartzentruber J, Abu Khadija K, Zaineddin S, Gamal H, Majewski J, Ben-Omran T. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin Genet. 2014 Aug;86(2):134-41. DOI: 10.1111/cge.12280
[70] Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014 Feb 13;506(7487):185-90. DOI: 10.1038/nature12975.

